Risk-Based Approach to Medical Device Design & Manufacturing | Risk Management
Developing the Risk Management File
The “end product” of the ISO 14971 standard is the Risk Management File (RMF). The RMF is the collection of documents that describe how risks are evaluated and mitigated for a specific product. That means each product (or product family) needs its own RMF.
Typical documents that can be found in an RMF include:
- Risk Management Plan
- Hazard Analysis
- Design/Process FMEAs
- Risk Management Report
- Test Plans/Reports for Risk Controls
The RMF is a “living document”, meaning that it will be continually updated throughout the product development life cycle. Once the product hits the market, the post-market surveillance process, or “real-world” data collection, will provide valuable insights into how effective your risk controls are and if any modifications for safety are needed.
The backbone of your RMF is the risk management plan. It serves as your reference point for all risk management activities throughout the duration of the product life-cycle. The Risk Management Plan defines the following for a product’s risk management process.
- Risk management activities
- Which activities need to occur in each design phase? Which activities will need to continue into the post-market phase?
- Required reviewers for each risk management activity
- Who must conduct the review? What does the review consist of? Any approvals that are required.
- Risk acceptability criteria
- What levels of risk are acceptable? How will acceptability be determined if a quantitative probability cannot be assigned to the hazard?
- Residual risk acceptance criteria
- What level of residual risk as acceptable? How will residual risk be identified and determined?
- Plan for verification of risk controls
- How will it be verified that risk controls have been implemented into the design? How will these controls be reviewed to determine effectiveness at mitigating risk?
- Plan for collecting and reviewing post-production information
- What sources will be used to gather post-production data? How will that data be reviewed? How will it be used as an input to the ongoing risk management process?
The risk management process can be presented as a 6-step process.
Phase 1: Risk Analysis Stage
During this stage, you will identify characteristics related to safety, based on your device’s intended use and reasonably foreseeable misuse, and then estimate the risk for each hazard. ISO 14971 defines reasonably foreseeable misuse as “use of a product or system in a way not intended by the manufacturer, but which can result from readily predictable human behavior.” Hazards are not the same as hazardous situations.
A hazard is defined as a potential source of harm, and a hazardous situation is the circumstance in which people, property, or the environment is/are exposed to one or more hazards. Thus, a hazard can lead to a hazardous situation, which can cause harm. To estimate the risk of a hazard, you will need to assess the probability of the situation’s occurrence (ranging from Improbable to Frequent) and the severity of the harm (ranging from Negligible to Catastrophic). This can be done by referencing academic/clinical publications, similar device data, and reliably published public data.
Phase 2: Risk Evaluation
In this phase, you will determine if each risk is acceptable or not. Risk acceptability is defined in your risk management plan for the product. The graphic below is an example of how the combination of probability and severity are used to determine if a hazard poses insignificant or unacceptable risk. Based on your company’s specific risk acceptance policy, hazards that result in insignificant risk are often not required to be mitigated (i.e. no risk controls needed).
Hazards that result in unacceptable risk or call for further risk reduction are required to have risk controls in place to mitigate the risk. Risk control is the process by which risks are reduced or maintained to a specified level, and is documented in your risk management plan.
Severity of Hazard Table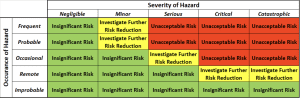
Phase 3: Risk Control
Once you identify which risks need to be controlled, you must use one or more of the risk control options included in ISO 14971:
- Inherently safe design
- Protective measures
- Information for safety & training
If you find that you can’t reduce a risk to an acceptable level with any of these risk control options, you must generate a benefit-risk analysis to justify the risk. If the benefit does not outweigh the risk, it is recommended to go back to the three risk control options and, in an iterative process, implement one or more controls until the residual risk is acceptable. Once you identify your risk controls, you must implement and verify that the controls effectively reduced the risk within the acceptable range.
After the residual risk is deemed acceptable, you must consider if any new hazards or hazardous situations arise due to implementing your risk controls. If so, return to the risk evaluation stage and document these new hazards or hazardous situations. Once all hazardous situations are considered, you are ready for the next phase of the risk management process: overall residual risk.
Phase 4: Evaluation of Overall Residual Risk
In this phase, you consider all your risks and determine if the benefits outweigh all of these risks combined. Your acceptability criteria should be documented in your risk management plan. If you find that the overall residual risk is unacceptable, you will need to go back through the entire risk management process to make modifications or consider changing your device’s intended use.
Phase 5: Risk Management Review
At this point, you have identified, evaluated, and controlled your risks. You have deemed the device beneficial despite these risks, and you are preparing for your device to go to market. Your team shall review and approve that the risk management file is complete. The review must be documented in a risk management report.
Phase 6 and Ongoing: Production and Post-Production Activities
Once your risk management file is “completed”, you will need to identify sources of production and post-production activities, which will feed back into your product’s risk. This may include information from non-conformances, CAPAs, complaints, or information from publicly available databases, including the FDA’s MAUDE (Manufacturer and User Facility Device Experience) database. These sources of data will help you refine your device and challenge you to make modifications to your device, keeping it safe for your intended users.