Design & Process FMEA (Failure Modes and Effects Analysis) | Risk Management
ISO 14971 is the international standard that outlines the risk management process for medical device manufacturers. The standard requires the use of risk management tools to analyze and evaluate risks throughout the device lifecycle. The minimum required tool is the hazard analysis – a top-down technique where the product team identifies all possible hazardous situations during device use or manufacturing or assembly process. This process focuses on the consequences on the patient or user.
A Failure Modes and Effects Analysis (FMEA) can complement this standard as a risk assessment tool. FMEA is a qualitative “bottom up” technique that assists in the identification and evaluation of causes of failures and their effects on the product and end user. Each component is evaluated one at a time for different ways in which the component/product can fail to perform its intended function throughout its life cycle. Simbex uses the FMEA technique to identify both failures of the device and potential hazards that could result in harm. We implement an FMEA for every design and development project we do, and every client receives thorough and concise documentation for their mandated risk management file. The two most common types of FMEAs are DFMEAs (Design FMEAs) and PFMEAs (Process FMEAs). These two FMEAs are complementary and vital to developing a robust risk management file.
Medical Device Design FMEA
In DFMEAs, you assess the potential failures of components in your product’s design. You will consider each component of your device and how it functions, then assess the likelihood of different failure modes.
For example, if you have a rechargeable, wearable medical device with a LED indicator that lights up when the device is charging, your DFMEA may include a scenario in which the indicator does not light up once it is plugged in to charge, or a scenario in which the indicator lights up when it is not charging. Both of these scenarios are unintended failures and should be assessed. A potential mitigation (or design control) may include a secondary charging indicator or notification to users on the companion software application.
It is advised to begin your DFMEA in conjunction with the beginning stages of your design and development process. This allows for any design controls to be implemented in the design input stage which expedites your design process, allowing your team to catch weaknesses or flaws in your device before design freeze and design transfer.
In the long run, the DFMEA will save you money and reduce your product development time and maintain the quality of your product.
Benefits of PFMEA in Production Processes for Medical Devices
In PFMEAs, you assess the potential failures during steps in your production processes and their effect on device quality. The purpose of the PFMEA is to identify areas where and how your process may fail, then implement strategies to mitigate those failures.
For instance, what could happen if an incorrect UDI barcode was printed on your product label? Perhaps the end user receives the wrong device, or traceability is compromised. To mitigate this failure mode from occurring (or process control), you could implement a barcode verification step during the manufacturing process to ensure that the correct UDI is printed on the label.
Just as you would for your DFMEA, it is beneficial to begin compiling your PFMEA early in the process development phase. This could occur concurrently with equipment or manufacturing process development and continue throughout process validation and beyond.
After design transfer, your PFMEA will continually undergo refinement in response to nonconforming materials and process errors, among other data sources.
In the long run, the PFMEA will help you maintain device quality throughout production.
Why Should I Perform an FMEA?
The FMEA process helps the development brainstorm the possible failure modes in the design or process in a straightforward manner and accelerates your design and development process through trial and error.
By anticipating failure modes in the premarket stage, you can design or change your device to reduce risk as much as possible before your device gets to market. Identifying failure modes before getting the device to user trials reduces design iterations and reduces the number of repeat user trials.
Once your device gets to market, you will likely observe some failure modes that you did not anticipate and a higher or lower incidence of a specific failure mode. These observations feed back into your product’s risk management file.
Thus, FMEAs are living documents and are continually updated throughout the product life cycle.
Tips for conducting an FMEA
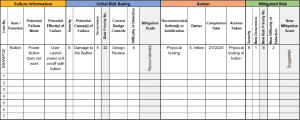
- To create an FMEA, it is important to have a diverse, cross-functional team to ensure that you are considering all aspects of the product or project. Your team will identify and document the various components of your design/process.
- Your team will identify the ways that the components of the design or process could fail.
- Once you define how the components could fail, you will brainstorm and document how the failure could affect the end user.
- A severity (S) score is given to the effect of the failure mode. These items are typically rated on a 1-10 scale and the scale rating definitions are adjusted to reflect the specific failure types of the device.
- Next, your team will identify the possible root causes of each failure mode, or the condition that could lead to failure. At this point, you have defined the failure, how the failure could happen, and how the failure impacts the end user.
- Then, estimate the occurrence (O) or probability of the root cause happening. These items are typically rated on a 1-10 scale and the scale rating definitions are adjusted to reflect the specific failure types of the device.
- Based on the pre-control Risk Priority Number (RPN) and your company’s risk acceptance policy, a design/process control is described to mitigate the failure mode.
- A detection (D) rating is applied to the implemented design/process control. The detection rating indicates the level of confidence that the design/process control will prevent the cause of the failure mode or detect the failure mode prior to releasing the product.
- The RPN is re-calculated, taking into account new risks associated with the implemented design/process controls.
- Once you’ve mitigated the risks of failure modes to an acceptable level per your company’s policy, the design/process FMEA can be considered complete for this version of the design/process.
- FMEAs are typically organized in a table, which allows you to easily add new rows of information and continually update your failure modes and effects.
The Simbex Advantage
Capturing all failure modes in your FMEA is a monumental task, requiring hours of effort from various team members. Risk assessment and analysis may be a requirement of the medical device product lifecycle, but the FMEA process rapidly pushes your product forward, and ultimately, helps get your product to market faster.
The risk management process is a long, winding road that will challenge how your device and processes function safely and effectively. Simbex’s 23+ years of experience and comprehensive interdisciplinary product development capabilities can help you overcome this hurdle by providing thorough and correct documentation of your design and process failure modes and effects analysis.